DRAGEN Methylation Pipeline
The epigenetic methylation of cytosine bases in DNA can have a dramatic effect on gene expression, and bisulfite sequencing is the gold standard for detecting epigenetic methylation patterns at single-base resolution. This technique involves chemically treating DNA with sodium bisulfite, which converts unmethylated cytosine bases to uracil, but does not alter methylated cytosines. Subsequent PCR amplification converts any uracils to thymines.
A bisulfite sequencing library can either be nondirectional or directional. For nondirectional, each double-stranded DNA fragment yields four distinct strands for sequencing, post-amplification, as shown in the following figure:
Nondirectional Bisulfite Sequencing
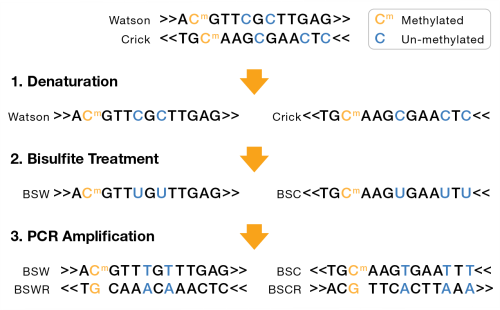
| – | Bisulfite Watson (BSW), reverse complement of BSW (BSWR), |
| – | Bisulfite Crick (BSC), reverse complement of BSC (BSCR) |
For directional libraries, the four strand types are generated, but adapters are attached to the DNA fragments such that only the BSW and BSC strands are sequenced (Lister protocol). Less commonly, the BSWR and BSCR strands are selected for sequencing (eg, PBAT).
BSW and BSC strands:
| • | A, G, T: unchanged |
| • | Methylated C remains C |
| • | Unmethylated C converted to T |
BSWR and BSCR strands:
| • | Bases complementary to original Watson/Crick A, G, T bases remain unchanged |
| • | G complementary to original Watson/Crick methylated C remains G |
| • | G complementary to original Watson/Crick unmethylated C becomes A |
Standard DNA sequencing is used to produce sequencing reads. Reads containing more methylated C’s or G’s complementary to methylated C’s are less drastically affected by the bisulfite treatment, and have a higher likelihood of mapping to the reference than reads with more bases affected. The standard protocol to minimize this mapping bias is to perform multiple alignments per read, where specific combinations of read and reference genome bases are converted in-silico prior to each alignment run. Each alignment run has a set of constraints and base-conversions that corresponds to one of the bisulfite+PCR strand types expected from the protocol. By comparing the per read (pair) alignments across runs, DRAGEN determines the best alignment, and most likely, strand type for each read or read pair. DRAGEN can then use the alignments and strands for downstream methylation calling.